Research
Three-dimensional genome organization plays an essential role in all DNA-templated processes, including gene transcription, regulation, replication, etc. Computational modeling can serve as an effective way of building high-resolution genome structures and improving our understanding of these molecular processes. While in silico studies of protein folding have become a rather mature field, simulating the human genome faces significant challenges because of its enormous size (over 6 billion base pairs), the complexity of molecular players involved in its 3D organization, and the non-equilibrium nature of cell nuclei. We tackle these challenges by bringing together statistical mechanical theory, computer simulations, and machine learning to invent new methodologies and model the genome at different length scales with chemical specificity.

Chromatin folding with atomistic details
In one research direction, we are characterizing genome organization at a near atomistic resolution. Our simulation approach generalizes beyond existing phenomenological models by providing a vastly improved description of protein-protein and protein-DNA interactions. It is helping us resolve the long-standing controversy regarding the most stable organization for a string of nucleosomes and the existence of regular 30 nm fibers in situ. Our innovation here goes into the development of force fields and sampling techniques to ensure both chemical accuracy and computational efficiency of chromatin modeling. These two features are essential for systematically investigating the dependence of chromatin stability on ionic concentrations, nucleosome repeat length, DNA sequence, and histone modifications.
Whole genome organization with integrative modeling
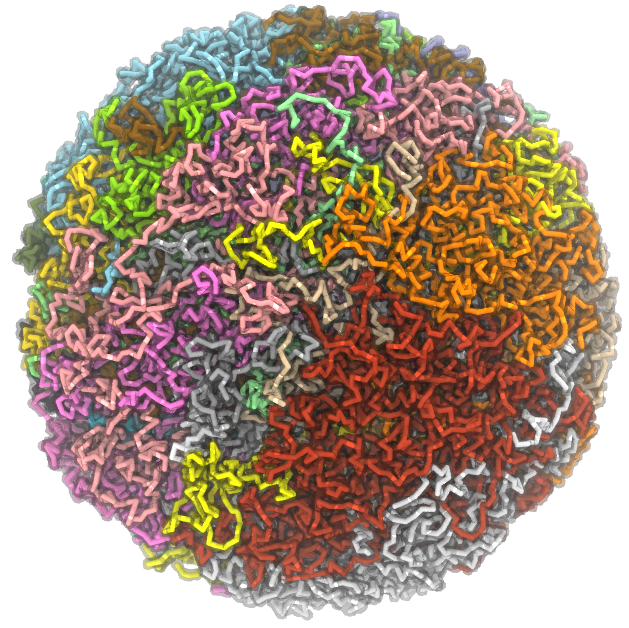 Due to its high computational cost, the first-principles approach outlined above will be limited to study a single gene. To simulate the whole genome consisting of over 20,000 genes, we are developing computational approaches inspired by statistical mechanics, polymer simulations, and recently deep learning. One particular direction is what we call the data-driven mechanistic-modeling approach, which enables a high-resolution reconstruction of three dimensional genome organization. Importantly, it makes possible mechanistic studies to investigate casual relationships between genome organization and genetic and epigenetic marks. Meanwhile, we are working closely with experimental groups to identify crucial molecular players for organizing the genome and to investigate the implications of genome misfolding in tumorigenesis.
Due to its high computational cost, the first-principles approach outlined above will be limited to study a single gene. To simulate the whole genome consisting of over 20,000 genes, we are developing computational approaches inspired by statistical mechanics, polymer simulations, and recently deep learning. One particular direction is what we call the data-driven mechanistic-modeling approach, which enables a high-resolution reconstruction of three dimensional genome organization. Importantly, it makes possible mechanistic studies to investigate casual relationships between genome organization and genetic and epigenetic marks. Meanwhile, we are working closely with experimental groups to identify crucial molecular players for organizing the genome and to investigate the implications of genome misfolding in tumorigenesis.
Non-equilibrium chromatin dynamics
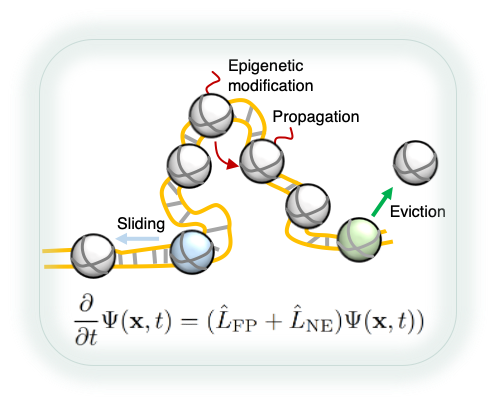 Chromatin is inherently an active system. Its conformational dynamics is subject to perturbations from ATP-driven enzymes that break the detailed balance. In addition, post-translational modifications to histone proteins undergo constant exchange, resulting in a time-dependent Hamiltonian. We are developing analytical approaches to address the impact of non-equilibrium activities on chromatin organization, with a focus on perturbation theories that approximate non-equilibrium steady state with effective equilibrium models.
Chromatin is inherently an active system. Its conformational dynamics is subject to perturbations from ATP-driven enzymes that break the detailed balance. In addition, post-translational modifications to histone proteins undergo constant exchange, resulting in a time-dependent Hamiltonian. We are developing analytical approaches to address the impact of non-equilibrium activities on chromatin organization, with a focus on perturbation theories that approximate non-equilibrium steady state with effective equilibrium models.
Advancing molecular simulations with deep learning
 Molecular simulations are powerful techniques that characterize systems of interest at a detailed level that is hard to achieve experimentally. However, to fully unleash the power of molecular simulations, several aspects need to be further improved. There is a growing need for developing accurate coarse-grained models for long-timescale simulations of large systems. In addition, a central quantity actively sought after in computational studies is the free energy of a state. While numerous techniques have been introduced for its computation, they often struggle to balance efficiency and accuracy. Harnessing the remarkable progress in deep learning and generative modeling, we aim to significantly improve molecular simulations’ efficiency and quality.
Molecular simulations are powerful techniques that characterize systems of interest at a detailed level that is hard to achieve experimentally. However, to fully unleash the power of molecular simulations, several aspects need to be further improved. There is a growing need for developing accurate coarse-grained models for long-timescale simulations of large systems. In addition, a central quantity actively sought after in computational studies is the free energy of a state. While numerous techniques have been introduced for its computation, they often struggle to balance efficiency and accuracy. Harnessing the remarkable progress in deep learning and generative modeling, we aim to significantly improve molecular simulations’ efficiency and quality.
Multiscale simulation of biomolecular condensates
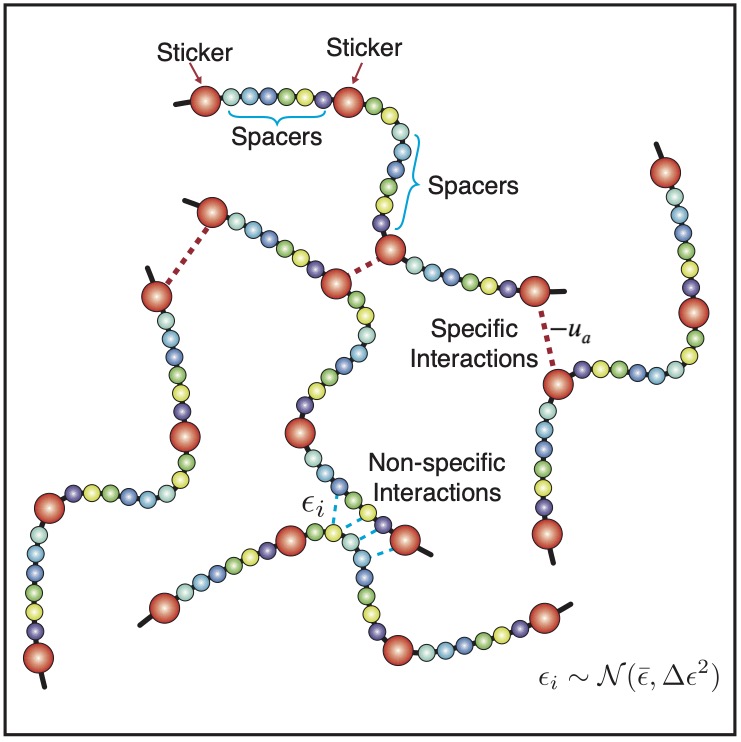 Many membraneless organelles, or biological condensates, form through phase separation, and play key roles in signal sensing and transcriptional regulation. While the functional importance of these condensates has inspired many studies to characterize their stability and spatial organization, the underlying principles that dictate these emergent properties are still being uncovered. We develop analytical theory and multiscale simulation techniques to elucidate the “molecular grammar” that connects amino acid sequences with protein phase behaviors and the collective physical properties of condensates.
Many membraneless organelles, or biological condensates, form through phase separation, and play key roles in signal sensing and transcriptional regulation. While the functional importance of these condensates has inspired many studies to characterize their stability and spatial organization, the underlying principles that dictate these emergent properties are still being uncovered. We develop analytical theory and multiscale simulation techniques to elucidate the “molecular grammar” that connects amino acid sequences with protein phase behaviors and the collective physical properties of condensates.