Research
We develop machine learning and artificial intelligence–driven multiscale simulation technologies to explore how molecular interactions give rise to biological organization. By combining theoretical chemistry, statistical mechanics, and data-driven modeling, our work extends the reach of chemistry into complex biological systems—allowing us to apply chemical principles to study processes that span from atomic interactions to cellular organization. These technologies enable chemically informed simulations of genome architecture, providing a mechanistic link between molecular structure and biological function. This chemistry-centered approach offers a powerful framework to uncover how physical interactions and molecular composition shape gene regulation, with potential implications for genome engineering and therapeutic discovery. Beyond chromatin organization, our methods are broadly applicable to other multicomponent biomolecular systems, such as condensates and dynamic macromolecular assemblies, where molecular details are essential to understanding emergent biological behavior.
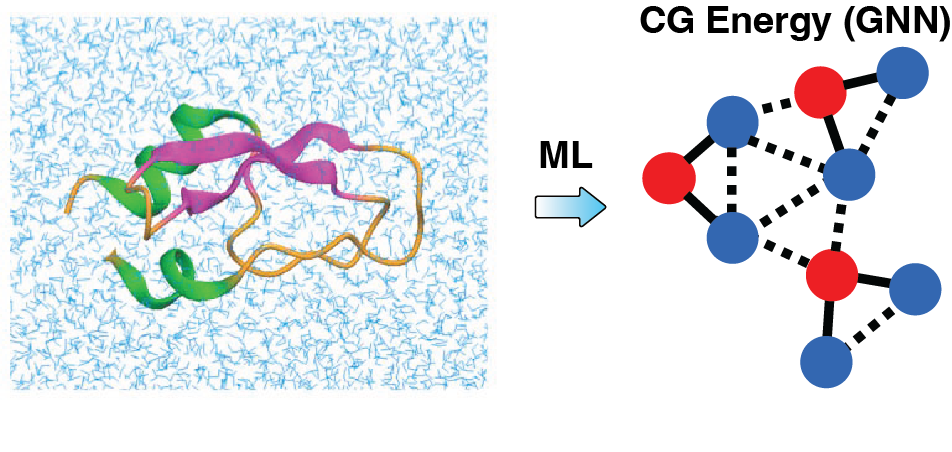
Machine Learning–Based Coarse Graining
Traditional coarse-grained (CG) models have been instrumental in extending the reach of molecular simulations to larger systems and longer timescales. However, their accuracy has often been limited by simplified energy functions and the lack of systematic optimization frameworks. Our group is advancing this field by combining machine learning with principles of statistical mechanics to develop CG models that are thermodynamically consistent with atomistic simulations and rigorously reproduce configurational statistics. Leveraging data from high-resolution simulations, we design optimization algorithms that directly enforce physical consistency across resolutions. To represent complex many-body interactions with chemical specificity, we have introduced graph neural network (GNN) architectures tailored for molecular energy landscapes. These architectures allow flexible yet physically interpretable representations of coarse-grained potentials. By integrating these approaches, we are building a next generation of coarse-grained models that retain the efficiency of reduced representations while achieving unprecedented accuracy and transferability across molecular systems. These developments are enabling simulations that connect chemical details to mesoscale organization and emergent biological function.
Bridging Mesoscale and Near-Atomistic Modeling of Chromatin Organization
 Understanding how chromatin folds to regulate gene activity requires connecting structural organization across vastly different length scales. We are integrating data-driven mesoscale chromatin modeling with physics-based near-atomistic simulations to unravel the molecular mechanisms of chromatin folding at the gene scale. Our mesoscale models reconstruct biologically relevant chromatin configurations at nucleosome resolution, enabling quantitative exploration of large genomic domains. In parallel, our near-atomistic simulations explicitly resolve individual amino acids and nucleotides, capturing the physicochemical interactions and structural motifs that govern chromatin dynamics.
Understanding how chromatin folds to regulate gene activity requires connecting structural organization across vastly different length scales. We are integrating data-driven mesoscale chromatin modeling with physics-based near-atomistic simulations to unravel the molecular mechanisms of chromatin folding at the gene scale. Our mesoscale models reconstruct biologically relevant chromatin configurations at nucleosome resolution, enabling quantitative exploration of large genomic domains. In parallel, our near-atomistic simulations explicitly resolve individual amino acids and nucleotides, capturing the physicochemical interactions and structural motifs that govern chromatin dynamics.
By combining these two complementary approaches, we aim to achieve continuous chromatin representations at sub-nanometer resolution, bridging nucleotide-level detail with large-scale genome architecture—an unprecedented integration in the field. These simulations will provide molecular insights into the driving forces that mediate enhancer–promoter communication and the mechanisms that control gene regulation within the three-dimensional genome.
Generative AI for Genome Sequence–Structure–Function Relationships
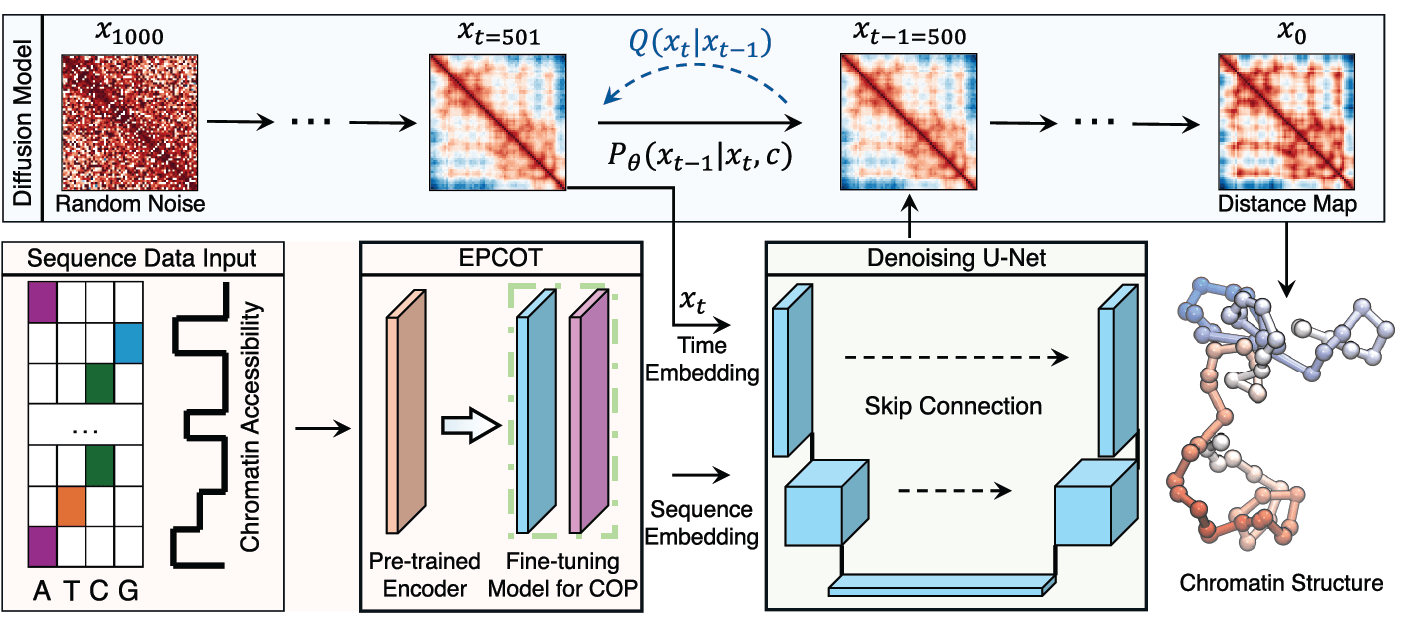 In parallel with our physics-based modeling efforts, we are harnessing the power of generative artificial intelligence to explore how genome sequence encodes three-dimensional structure and biological function. Generative AI models excel at capturing contextual dependencies within complex data. Much like large language models that learn the rules of human language, these models can be trained to decode the grammar of the genome — identifying how sequence patterns give rise to structural and regulatory features.
In parallel with our physics-based modeling efforts, we are harnessing the power of generative artificial intelligence to explore how genome sequence encodes three-dimensional structure and biological function. Generative AI models excel at capturing contextual dependencies within complex data. Much like large language models that learn the rules of human language, these models can be trained to decode the grammar of the genome — identifying how sequence patterns give rise to structural and regulatory features.
By learning these sequence–structure–function relationships, generative models provide a new means to predict chromatin organization directly from DNA sequence and to uncover the molecular code underlying gene regulation. Moreover, these models can be used in a creative capacity to generate de novo genomic sequences with desired structural or functional properties, opening new avenues for rational genome engineering and therapeutic design.
Multiscale simulation of biomolecular condensates
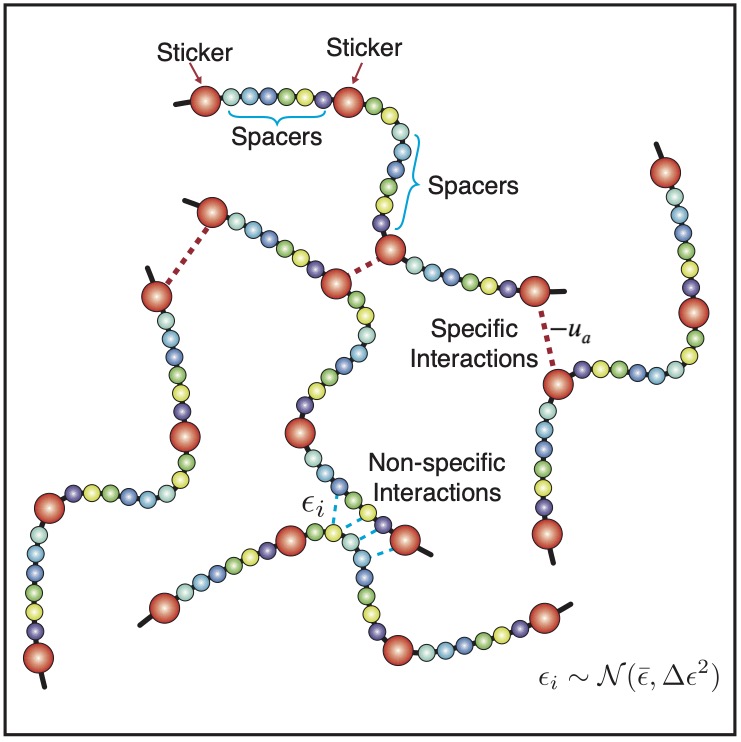 Many membraneless organelles, or biological condensates, form through phase separation, and play key roles in signal sensing and transcriptional regulation. While the functional importance of these condensates has inspired many studies to characterize their stability and spatial organization, the underlying principles that dictate these emergent properties are still being uncovered. We develop analytical theory and multiscale simulation techniques to elucidate the “molecular grammar” that connects amino acid sequences with protein phase behaviors and the collective physical properties of condensates.
Many membraneless organelles, or biological condensates, form through phase separation, and play key roles in signal sensing and transcriptional regulation. While the functional importance of these condensates has inspired many studies to characterize their stability and spatial organization, the underlying principles that dictate these emergent properties are still being uncovered. We develop analytical theory and multiscale simulation techniques to elucidate the “molecular grammar” that connects amino acid sequences with protein phase behaviors and the collective physical properties of condensates.